Home
![]()
💫 Graph-based foundation model for spatial transcriptomics data
Novae is a deep learning model for spatial domain assignments of spatial transcriptomics data (at both single-cell or spot resolution). It works across multiple gene panels, tissues, and technologies. Novae offers several additional features, including: (i) native batch-effect correction, (ii) analysis of spatially variable genes and pathways, and (iii) architecture analysis of tissue slides.
Overview
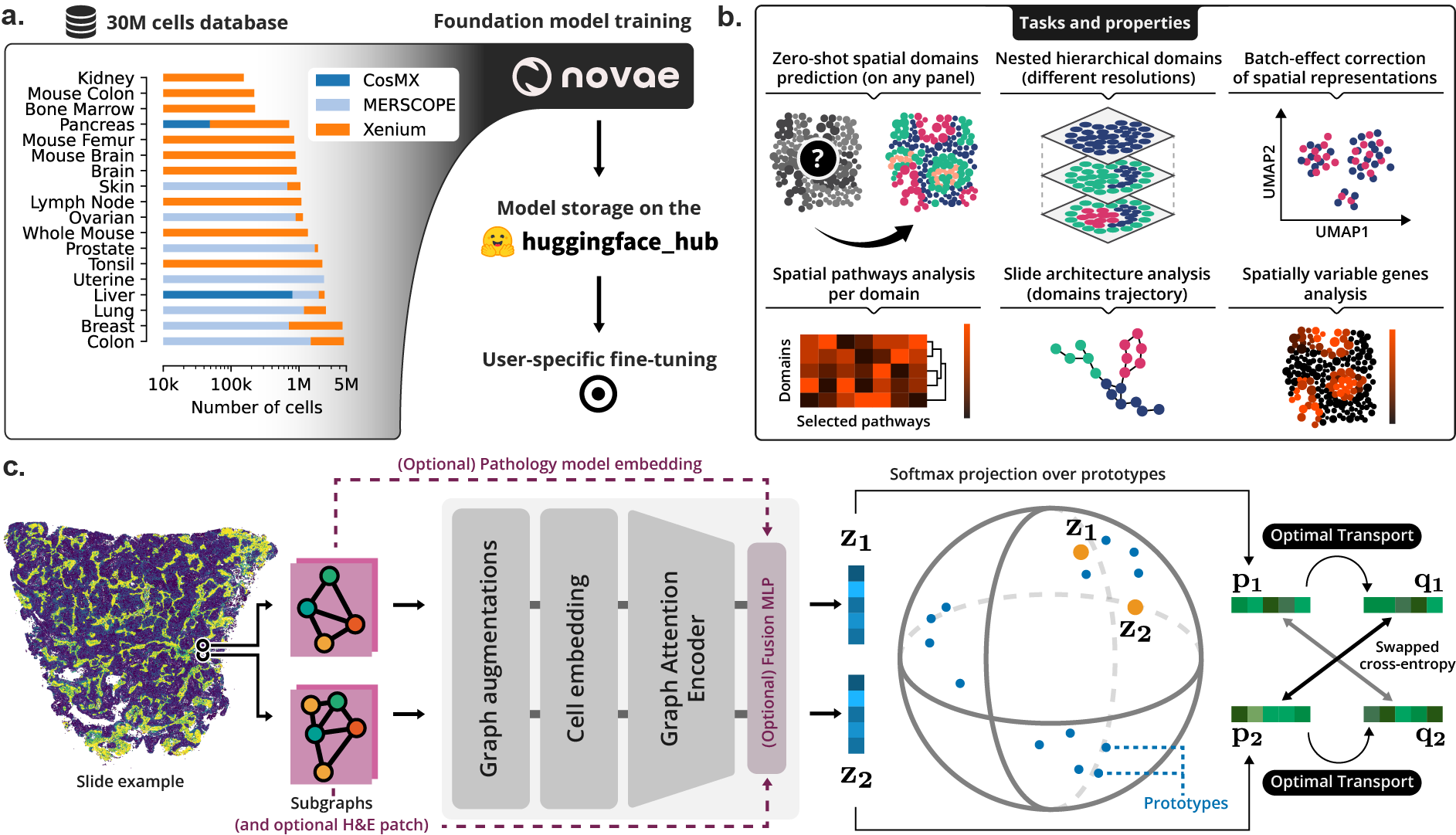
(a) Novae was trained on a large dataset, and is shared on Hugging Face Hub. (b) Illustration of the main tasks and properties of Novae. (c) Illustration of the method behind Novae (self-supervision on graphs, adapted from SwAV).
Why using Novae
- It is already pretrained on a large dataset (pan human/mouse tissues, brain, ...). Therefore, you can compute spatial domains in a zero-shot manner or quickly fine-tune the model.
- It has been developed to find consistent domains across many slides. This also works if you have different technologies (e.g., MERSCOPE/Xenium) and multiple gene panels.
- You can natively correct the spatial-domains batch-effect, without using external tools.
- After inference, the spatial domain assignment is super fast, allowing you to try multiple resolutions easily.
- It supports many downstream tasks, all included inside one framework.